-

-
上海沙格企业管理咨询有限公司
FDA验厂,欧盟自由销售证书,医疗器械单一体系审核MDSAP,CE第四版临床评价报告,MDR CE认证 - 咨询热线:袁小姐
13818104617




热门搜索:

2017年2月Regulation (EU) 2017/745 on Medical Devices医疗器械法规(MDR)提案发布,同年3月,欧盟成员国一致表决同意MDR。2017年5月5日,欧盟Official Journal正式对外宣MDR法规内容。
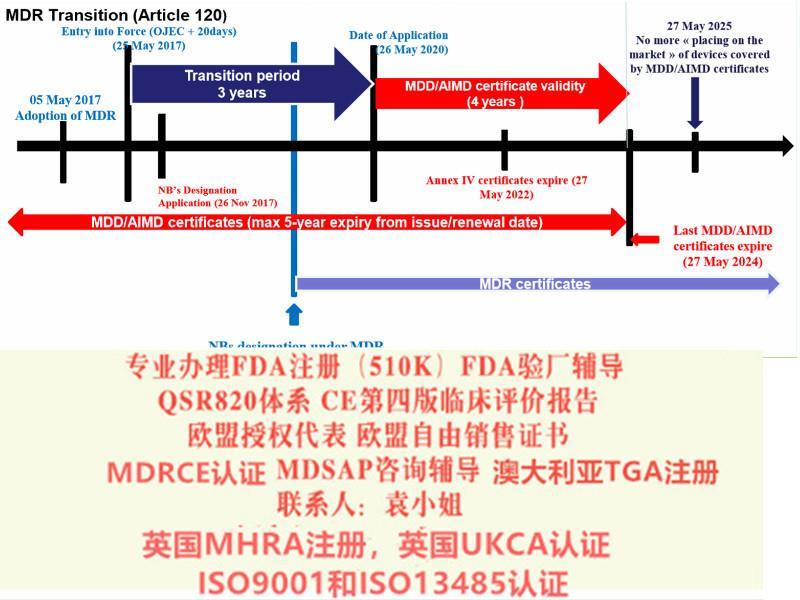
MDR新法规将取代现行的有源医疗器械指令Council Directive 90/385/EEC on Active Implantable Medical Devices (AIMDD) (1990)以及医疗器械Council Directive 93/42/EEC on Medical Devices (MDD) (1993)指令。原计划2020年5月26日正式实施的MDR受**新冠影响将推迟实施时间至2021年5月26日。
在此期间,仍然可以进行以下MDD证书的相关活动,例如产品变更及一年期内MDD新户的申请;现有MDD客户证书更新的相关活动;现有MDD客户证书更新的申请(包括提前更新的申请);现有MDD客户重大变更的申请期。虽然MDR的正式实施有延期,但是有例外情况如:
· MDD产品的投放截止日期仍为2024年5月26日
· MDD产品供应截止日期仍为2025年5月26日。
对制造商和产品的影响而言,93/42 EEC指令和MDR基本上具有相同的基本监管要求。没有删除现有的需求,但是MDR添加了新的需求,与目前93/42指令相比,MDR更加强调生命周期方法的*性,并有临床数据支持。MDR对公告机构的*提出了更严格的要求,对国家主管当局和**加强了控制和监测。MDR对某些设备重新分类,范围更广。例如,MDR明确涵盖用于清洁、消毒或消毒其他医疗设备的所有装置(第2.1条);一次性医疗器械(第17条)};和某些无医疗用途的装置(附件十六)。MDR还包括在互联网上销售医疗器械以及用于远程提供诊断或**服务的医疗器械(第6条)。MDR为一些Ilb类器械和植入性Il类器械引入了由独立*小组进行的临床评估咨询程序(第54条)。
新的标识识别系统(UDI系统)(第27条)将有力地增强市场后*相关活动的可追溯性和有效性。
MDR还将提高透明度,公开有关设备和研究的信息。新的欧洲医疗设备数据库eudamed将在提供数据和增加数据的数量和质量方面发挥核心作用(第33条)
2021年MDR法规的实施日期逐渐临近,如何在过渡期截至前取得MDD的证书,如何或快速取得MDR证书。上海沙格有着的咨询师团队,丰富的案例经验。
根据欧洲法规的要求,制造商应起草符合性声明,对于加贴CE标志上市的产品的符合性负有责任。因此,制造商在产品进入欧洲市场前,需要评估并选定适用的指令和符合性路
径,确保产品符合法规要求,然后加贴CE标志。其主要过程包括:
1. 确定根据指令的定义,产品是否属于医疗器械,哪个欧盟指令适用于所考虑的医疗器械:医疗器械指令(93/42 / EEC),体外诊断器械指令(98/79/EC)或有源植入式医疗器械指令(90/385 / EEC)
2. 确定医疗器械的分类。
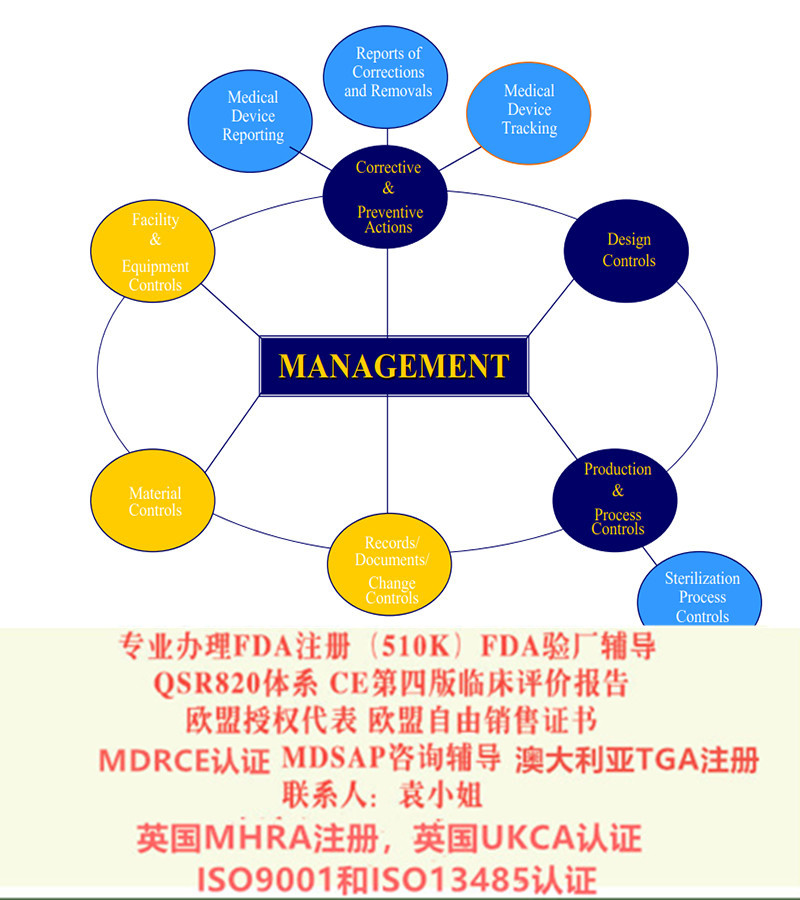
3. 实施质量管理体系(如适用)。大多数公司使用ISO 13485来满足要求。
4. 准备CE标记的技术文件(Technical Documentation)或设计文档(Design Dossier)。
5. 如果制造商在欧洲没有实际的场地,需要任命一位欧洲授权代表,代表制造商在欧盟境内进行相关的法规联络事宜。
6. 制造商的质量管理体系(QMS)和技术文件/设计文档由公告机构(Notified Body)审核和评估。若器械属于I类非灭菌、I类无测量功能或顾客定制产品,则不需要公告机构的介入。
7. 从公告机构处获得CE证书和ISO 13485证书。
8. 起草符合性声明(DoC),其中所涉及的医疗器械符合相应的指令。
9. 在产品上(包括标签、说明书等)加贴CE标志,并投放欧洲市场。对部分欧洲国家,可能需要在上市前在该国医疗器械主管当局处进行备案。
10. 维持质量体系和CE证书的有效性。
我司可以办理:
1:欧盟MDR CE,欧盟授权代表,欧盟注册
2:美国FDA注册,FDA510K
3:国内的医疗器械注册证和生产许可证
4:加拿大的MDEL注册
5:ISO13485咨询和认证