办理加拿MDEL注册需要什么资料?-需要什么材料
更新时间:2024-04-28 浏览数:578
所属行业:
咨询 管理咨询
发货地址:上海市金山区石化街道
产品规格:2
产品数量:2345.00个
包装说明:3
价格:面议
产品规格2包装说明3咨询范围2
咨询类型4
服务项目3
品牌4
什么是CMDCAS?CMDCAS是The Canadian Medical Devices Conformity Assessment System(加拿大医疗器械格评定体系)的简写。经过2001年7月1日到2002年12月31日的过渡时期后,2003年1月1日强制执行,依据2001年7月4日医疗器械法令第97(3)修正案要求,加拿大健康部推行VIP过渡期。法令第97(3)原来要求于2001年7月1日开始强制执行医疗器械法令关于质量管理体系的要求,但是因为修正案的出台,量管理体系的要求推迟于2003年1月1日强制执行。加拿大卫生部(Health Canada)要求所有入加拿大市场销售的医疗器械制造商要CMDCAS(加拿大医疗器械合格评定体系)认证注册的认证证书,用以证明符合加拿大的医疗器械法规。
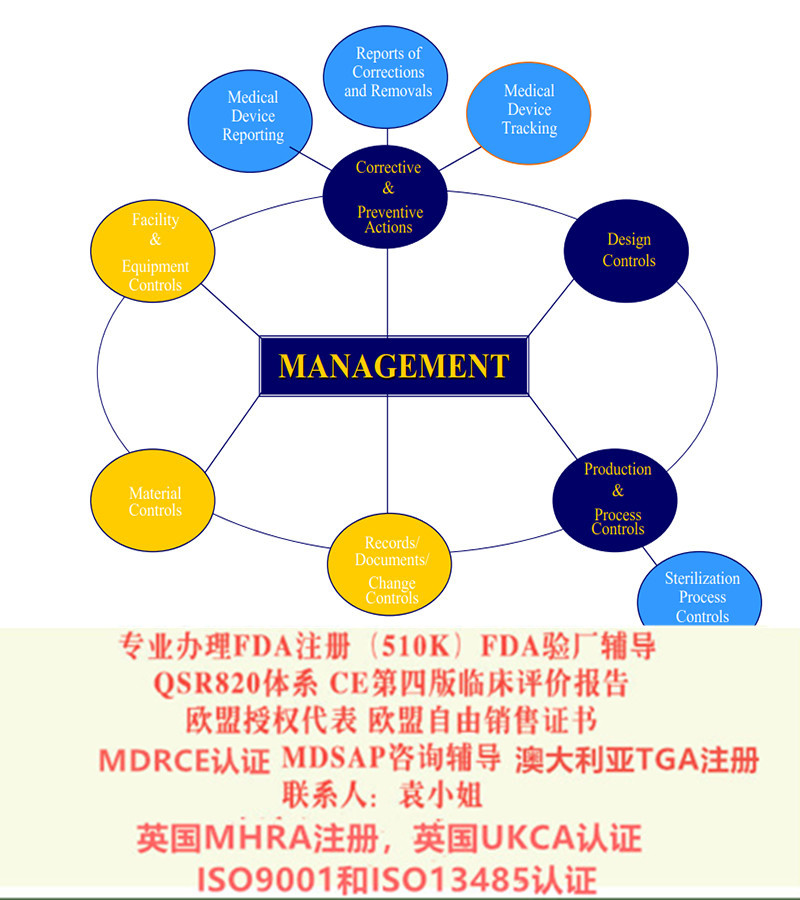
加拿大的医疗管理实行产品注册制度。不同于美国食品药品管理局(FDA)**彻底管制,由**对产品注册管理,再由**的现场审(GMP审查),亦不同于欧洲的完全第三方公告机构(Notified Body)检查制度(CE 认证),加拿大实行**注册结合第三方的质量体系审查。这里所说的第三方,指经加拿大标准**(SCC) 所认可的能够进行加拿大医疗器械合格评定体系(CMDCAS, Canadian Medical Devices Conformity) 审核的第三方机构。CMDCAS注册及分类依据Canadian Medical Devices Regulations (CMDR) SOR/98-282 as published by Health Canada.根据器械的使用风险将医疗器械分为I, II,III,IV四个分类,如I类器械为风险,IV类器械风险为。为此针对制造者提出的产品注册要求也是逐级增加,要求制造者实行的体系是愈加详尽。注册的基本流程介绍如下:
Class I:1. 为申请Medical Device Establishment License (MDEL)准备相应的技术文件2. 提交MDEL申请,支付卫生部行政收费。3. 申请评审通过,将在Health Canada网站公示。Class II:1. 通过CMDCAS认可的认证机构进行ISO 13485 审核认证(体系审核除ISO13485要求外还要包括CMDR的特殊要求),获得证书。2. 准备Canadian Medical Device License (MDL)申请。3. 提交MDL申请,并交纳卫生部行政收费。4. Health Canada评审MDL申请, 评审通过后进行网站公示。
Class III,IV:1. 通过CMDCAS认可的认证机构进行ISO 13485 审核认证(体系审核除ISO13485要求外还要包括CMDR的特殊要求),获得证书。2. 准备Canadian Medical Device License (MDL)申请。3. 提交MDL申请和Premarket review documents,并交纳卫生部行政收费。4. Health Canada评审MDL申请和Premarket review documents, 评审通过后进行网站公示。需要准备的文件清单I类医疗器械豁免注册。II,III,IV类器械的注册要求如下:1. 通用注册资料: a) 器械的名称; b) 器械的分类 c) 器械的标识; d) 产品标签上出现的制造者名称、地址; e) 若制造地点与 f) 不同,则制造地名称、地址;2. II 类器械注册附加资料: a) 所制造、销售或代理的器械关于医用条件的目的及用途的描述; b) 为满足*和有效性要求所符合的标准的清单; c) 由制造者的高层主管作的*有效性符合声明; d) 由制造者的高层主管作的器械标签符合加拿大医疗器械法规的声明; e) 若是近病人体外诊断设备(即不在医院而是在例如家庭使用的设备),制造者的高层主管应声明已用代表预期使用者的人体物质在与预期使用条件类似的条件下进行了研究性测试; f) 由CMDCAS认可机构颁发的MDSAP体系证书。
3. III类器械注册的附加条件: a) 器械及在其制造及包装中所用材料的描述; b) 所制造、销售和代理的器械在其允许的医疗条件、目的和用途下的性能描述; c) 除加拿大外的器械获准销售的国家清单、售出数量,以及报导的问题及召回情况 d) 器械的设计和制造为满足*有效性而采用的标准清单; e) 如果是以无菌出售的器械,则无菌方法描述; f) 制造者为*有效而进行的研究描述,以及由此得出的结论; g) 器械标签/复印件; h) 如果为近病人体外诊断设备,用代表目的预期用户的人体物质,以在使用条件类似的条件下的研究测试的情况; I) 所有与使用、*和有效有关的公开发布的报告的文献引用; j) 由加拿大医疗器械认证机构认可的机构所颁发的CAN/CSA-ISO 13485:984. IV 类器械注册的附加条件: a) 器械及制造和包装过程中所有材料的描述; b) 所制造、销售或代理的器械所允许的医疗条件、目的和用途的器械特性描述; c) 除加拿大以外器械获准销售的国家,售出数量,以及报告器械的问题及召回情况; d) 风险评估情况包括风险分析、风险评价,以及评价风险的满足*有效要求的措施; e) 与器械相关的质量计划,如特定的质量实践,资源及行动的程序; f) 制造和包装中使用的材料的参数; g) 器械的制造过程; h) 设计、制造中为满足*有效要求而采用的标准清单; I) 制造者为证明满足*有效要求而进行的所有研究的详细情况,包括: I) 临床前研究和临床研究; ii) 过程验证研究; iii) 适用时,软件验证研究,和 iv) 文献研究; j) 若非体外诊断设备、取自动物组织或组织衍生产物的器械,其客观生物*证据; k) 若为近病人体外诊断设备,针对代表预期用户的人体物质且在相似使用条件下进行的研究测试的详细情况; l) 制造者依据 I)款研究得出的结论; m) 制造者依据 h)款研究的总述及由此得出的结论; n) 与器械的使用、*和有效相关的公开发布报道的文献; o) 器械标签的复印件; p) 由加拿大医疗器械认证机构认可的机构所颁发的CAN/CSA-ISO 13485:98证书。医疗器械许可证发布后,每年11月1日应由制造者向加拿大卫生部提出再确认。取消生产许可证应在停止加拿大销售的30日之内提出。
http://sungofda.cn.b2b168.com